


COPYRIGHT © 广西百谷生物科技有限公司 ALL RIGHT RESERVED 桂ICP备18002610号-1 网站建设:中企动力 南宁
产品搜索:

广西百谷生物科技有限公司
公司名称:广西百谷生物科技有限公司
控股公司:广西晨农生物科技有限公司
办公地址:南宁市总部路3号中国-东盟科技企业孵化基地二期9栋3楼
扫一扫,关注我们
使用三代测序技术,精准解析菌群组成
浏览量
【摘要】:
广告位文本:
菌群组成解析,三代测序更精准!
导航栏关键字:
菌群组成谱测序
正文:
以PacBio公司的SMRT单分子实时测序技术(Single molecule real-time sequencing)为代表的三代测序技术具有无需PCR扩增富集、超长读长、碱基检测无偏好性和系统性偏差、一致性准确率高、原位实时直接检测DNA修饰等显著优势,已被证明能大幅改进微生物/动植物单个物种全基因组序列图谱的绘制能力,做到“零Gap”组装。那么,对于更为复杂的微生物组解析,三代测序的效果如何?

PacBio三代测序技术的主要优势
1.基于三代测序技术的多样性组成谱精细解析
在以16S/18S rRNA基因或ITS区域为目标序列的菌群多样性组成谱测序研究中,罗氏454、Illumina等二代测序平台一般只能对单个或连续的两三个可变区序列进行测序分析,通常情况下无法获得rRNA基因全长序列的信息,因而往往难以在属、种甚至菌株等精细水平识别测得的序列,难以获取“高分辨率”的微生物物种信息,菌群中大量成员对应的物种信息仍处于“模糊”状态,对于众多研究者而言无异于信息资源的“浪费”,是一个亟待攻克的难题。
而以PacBio公司的RS II和最新的Sequel测序系统为代表的三代测序技术,则有望从根本上一举解决上述困扰众多研究人员的心头之痛。首先,利用PacBio公司专利的SMRT单分子实时测序技术,可以对DNA序列实现单分子级别的超长读长测序,因而能够轻易读取微生物的rRNA基因全长序列;其次,通过PacBio所独有的环形一致性测序模式(Circular-consensus sequence,CCS),可以极大地提高单碱基测序的准确率,一般而言,将rRNA基因全长序列循环测序5~6次后,获得的一致性序列的准确率可以达到99.99%(QV40)以上,远远超过了二代测序的准确率,从而充分保障获得的rRNA基因全长序列的精确性。基于以上两点,PacBio的SMRT测序技术能够大大提高我们在种甚至菌株等精细水平解析菌群结构多样性的能力。
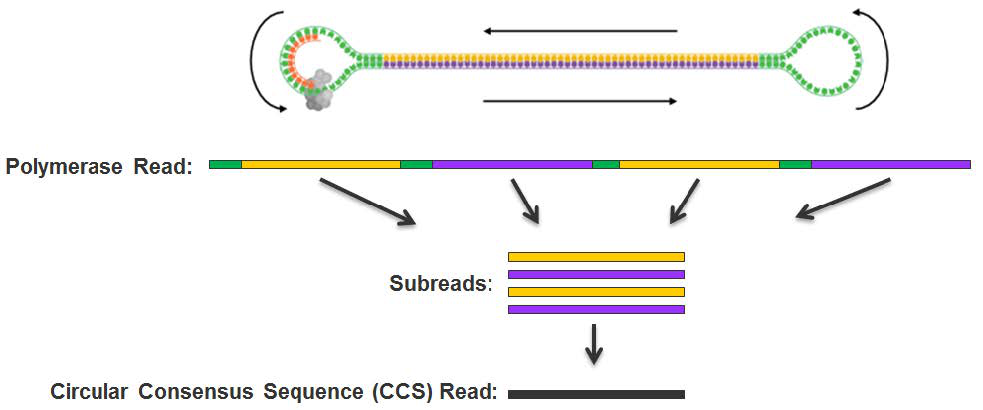
CCS测序模式基本原理
CCS测序模式能显著提高微生物rRNA基因全长的测序准确率
我们也把PacBio RS II和Illumina MiSeq的测序结果做过一些简单的比较分析:对同一个细菌群落样本的16S rRNA基因进行测序,在相同测序深度下,PacBio RS II的全长测序在种水平的分辨能力大大超过了Illumina MiSeq的短片段测序,使菌群组成得到精准解析;不仅如此,Illumina MiSeq会产生大量“无法确定归属”的物种,但这些物种的分类学信息都能通过PacBio RS II全长测序得到准确识别。
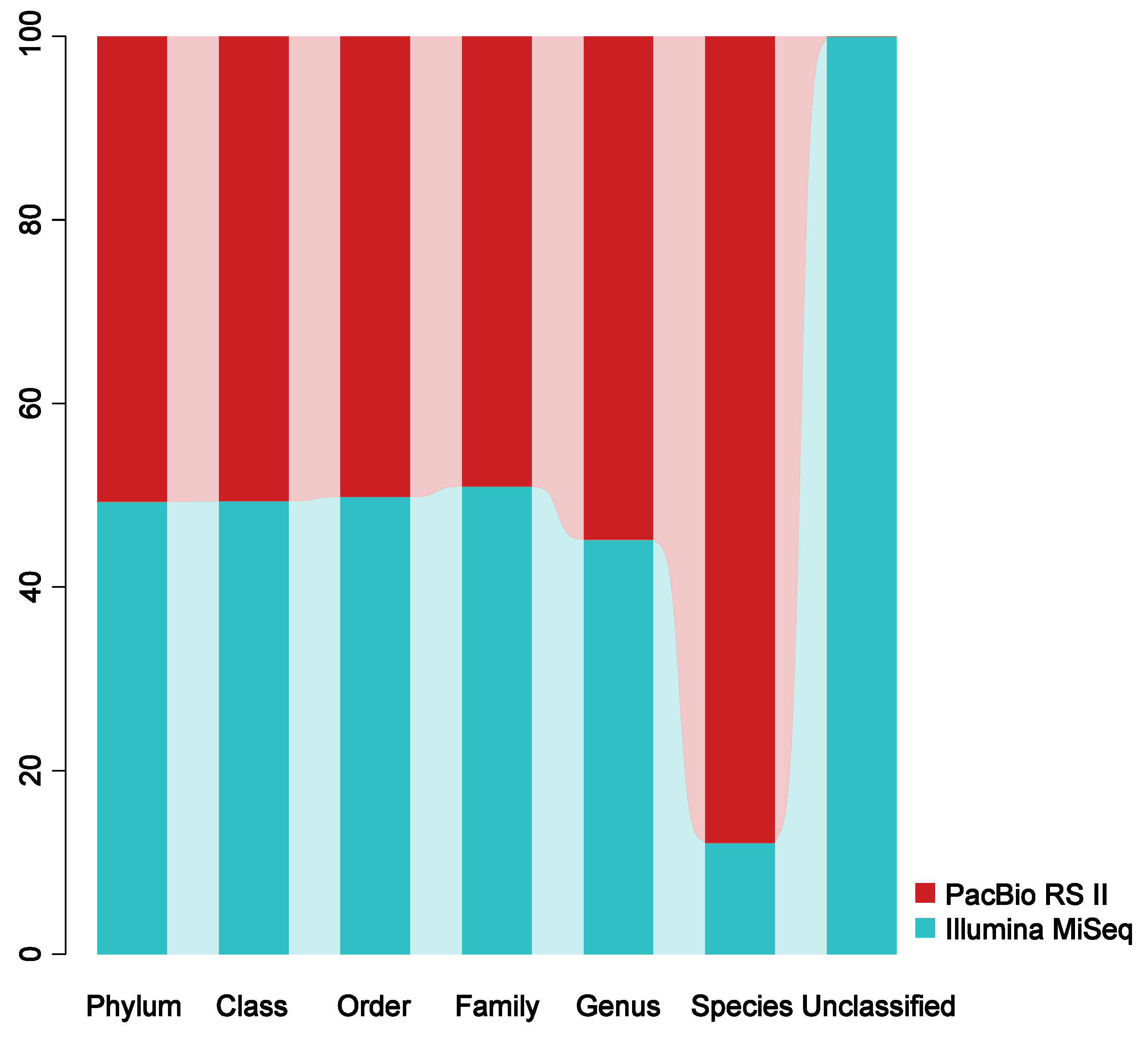
PacBio和Illumina平台对菌群16S rRNA基因测序结果的比较
2.案例解析
标题:利用三代测序技术实现对菌群多样性组成谱的“高分辨率”解析

原文链接:http://www.nature.com/ismej/journal/v10/n8/full/ismej2015249a.html
研究背景:
过去十多年中,随着短读长、高通量二代测序的兴起,以16S rRNA基因部分可变区序列为目标的分析方法,逐渐替代了基于rRNA基因全长克隆文库的一代Sanger测序法,使我们能对菌群组成进行深度定量解析。然而,由于二代测序读长短的特性,我们无法对测得物种进行精确的分类鉴定,从而限制了我们对菌群代谢功能的深入理解。随着三代SMRT测序技术的出现,我们有望从根本上解决这一瓶颈,实现对rRNA基因全长序列的高通量精准测序。
研究方法:
样本来源:23种细菌和3种古菌组成的人工菌群,以及取自湖水的天然微生物群落样本
测序平台:PacBio RS II(16S rRNA基因全长)+Illumina MiSeq(16S rRNA基因V4区)
对两种测序平台的测序结果进行多样性组成谱分析,并通过计算机模拟,比较16S rRNA基因全长序列和单V区序列的物种分辨能力。
研究结果:
根据两种平台的测序结果,门水平的菌群组成较为相似,但在更精细的水平存在差异。同时,三代测序的结果显著降低了物种分类信息的不确定性。计算机模拟的结果也表明,短读长的单V区测序可能严重低估某些特定类群的微生物,比如水体样本中参与氮循环和甲烷代谢的物种。因此,基于三代测序的菌群多样性组成谱分析能极大地提升物种分类鉴定的精确性,实现“高分辨率”检测的同时,也为深入阐释菌群的代谢功能奠定了基础。
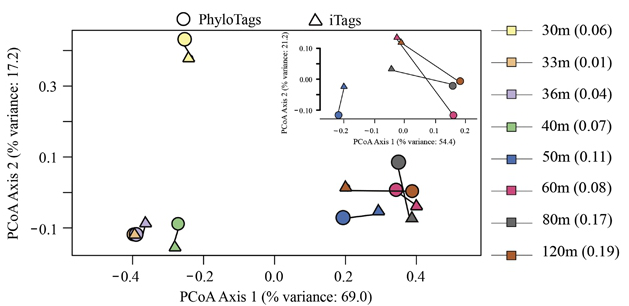
两种测序平台测序结果的整体比较。两者菌群的整体结构较为相似,但当菌群复杂程度增加时(子图),两者的检测结果有所差异。PhyloTags,三代PacBio RS II测序结果;iTags,Illumina MiSeq测序结果。
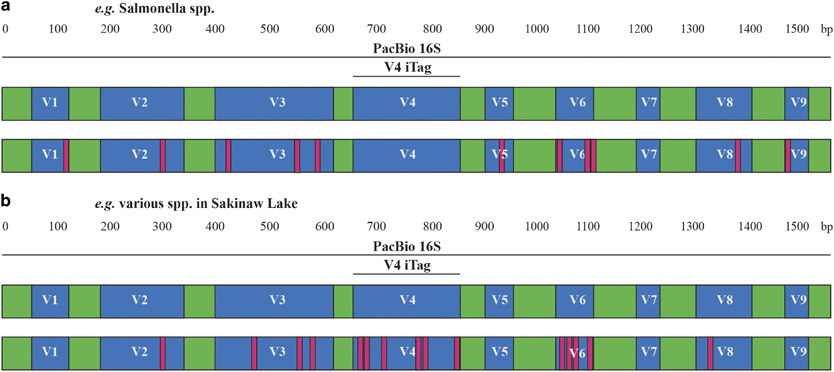
16S rRNA基因各V区的序列保守性并不一致,因而基于16S rRNA基因全长序列的三代测序,可以更全面地反映物种的种属信息。a图,以Salmonella属为代表,全长序列的种间差异达到97.4%,但V4区高度保守,二代测序结果将低估该物种的多样性;b图,某些物种在V4区的多样性可能高于其他V区,因而二代测序结果将高估相关物种的多样性。
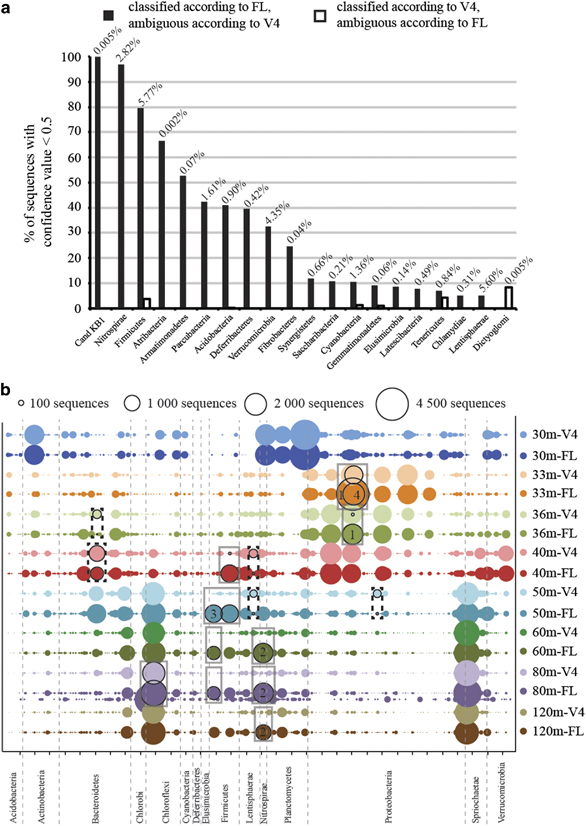
两种测序平台测序结果的精细比较。a图,三代测序的结果显著降低了物种分类信息的不确定性;b图,二代单V区测序结果可能会严重低估某些特定微生物类群的含量(以方框标记)。
研究结论:
综上所述,运用三代测序技术进行菌群多样性组成谱解析,将获得更为精准全面的结果。
3.派森诺优势
2016年,派森诺生物在原有的PacBio RS II三代高通量测序仪基础上,率先部署最新款Sequel测序仪,并已投入使用,独家提供16S rRNA基因全长和ITS全长测序分析服务,助力微生物组研究!

作为行业先锋,派森诺生物将一如既往地行使“解析序列,诠释生命”的理念,秉承“立足客户需要,满足个性需求”的服务宗旨,始终如一地提供性价比最高、最优质、最快速稳定的高通量测序和数据解析方案。
派森诺生物将竭诚为您服务!
参考文献
Singer, E., Bushnell, B., Coleman-Derr, D., Bowman, B., Bowers, R.M., Levy, A., Gies, E.A., Cheng, J.-F., Copeland, A., Klenk, H.-P., et al. (2016). High-resolution phylogenetic microbial community profiling. ISME J 10, 2020-2032.
上一篇:
无
简化基因组产品简介—广告文案
下一篇: